
Cos’è l’acromegalia?
L’acromegalia è una patologia endocrino-metabolica secondaria ad una esagerata secrezione di ormone della crescita; provoca una progressiva deformazione del volto, delle mani e dei piedi.
Come si manifesta?
Le mani e i piedi diventano più grandi: gli anelli non entrano più e diventano necessarie scarpe di due o tre numeri più grandi. Si verificano inoltre leggere ma evidenti trasformazioni anche nel viso: gli zigomi diventano sporgenti, le labbra si ingrossano, la mandibola si allarga tanto che tra dente e dente si creano degli ampi spazi.
Le trasformazioni sono molto lente e subdole. Inizialmente, il paziente acromegalico si sente molto forte fisicamente, pieno di energia e di voglia di fare; gli uomini, a volte, vedono migliorare le proprie prestazioni sessuali.
E’ difficile riconoscere in tutto questo dei segnali d’allarme di una malattia.
Dopo l’eccesso di benessere si determina una condizione di stanchezza generalizzata; frequentemente si osserva la scomparsa delle mestruazioni nella donna e una condizione di impotenza nell’uomo.
Se non curata l’acromegalia determina gravi conseguenze a livello metabolico (diabete mellito) e a carico di numerosi organi e apparati, con frequente comparsa di ipertensione, disturbi cardiaci, gravi difficoltà respiratorie, tumori dell’intestino, alterazioni tiroidee.
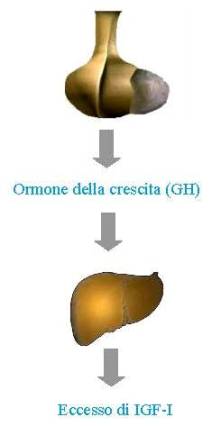
La causa dell’acromegalia è rappresentato da una eccessiva secrezione di ormone della crescita (GH, growth hormone) da parte di un tumore, solitamente benigno (adenoma), dell’ipofisi, una piccola ghiandola endocrina situata alla base del cervello.
L’ipersecrezione di GH determina a livello epatico e tissutale un’aumentata sintesi e secrezione di Insulin-like Growth Factor I (IGF-I), ormone che media gran parte degli effetti del GH. L’ipersecrezione di IGF-I è sostanzialmente responsabile della maggior parte dei segni e sintomi della malattia acromegalica.
L’acromegalia può colpire persone di ogni età, ma si riscontra più frequentemente negli adulti tra i 18 e i 30 anni.
Quando l’eccessiva produzione di GH si verifica prima che il processo di crescita sia ultimato, prima cioè della saldatura delle cartilagini epifisarie, si determina una crescita anomala di tutti i segmenti del corpo: tale condizione viene definita “gigantismo”.
La diagnosi di acromegalia
La diagnosi della malattia acromegalica è relativamente facile soprattutto nelle fasi avanzate.
La tipica facies acromegalica associata alle altre deformazioni devono indurre il sospetto di malattia, che deve essere dimostrata sul piano biochimico ed ormonale attraverso determinazioni, basali e dopo stimolo, di GH e IGF-I.
E’ necessario successivamente procedere ad una diagnosi di localizzazione tramite indagini radiologiche (TC, RMN), che generalmente permettono di dimostrare la presenza e l’estensione di un adenoma ipofisario.
A questa serie di indagini diagnostiche è ovviamente associata una estesa serie di indagini volte a definire le complicanze della malattia.
La terapia
La terapia degli adenomi ipofisari GH-secernenti può essere chirurgica, radioterapica o medica. Essa è finalizzata a:
a) rimuovere la neoplasia o ridurne il volume;
b) sopprimere l'ipersecrezione ormonale;
c) risolvere o almeno limitare insorgenza e progressione delle complicanze;
d) correggere eventuali deficit ipofisari associati con un adeguato trattamento sostitutivo.
 Attualmente, la terapia chirurgica rappresenta l’opzione terapeutica di prima scelta, soprattutto in presenza di microadenoma ipofisario (con diametro cioè, inferiore ad 1 cm). In questo caso, infatti, i risultati della neurochirurgia sono buoni, con percentuale di guarigione di circa 80%. In presenza di macroadenoma (evenienza assai più frequente) l’efficacia della terapia neurochirurgica è assai più modesta, con percentuale di guarigione mai superiore al 40% dei casi.
Attualmente, la terapia chirurgica rappresenta l’opzione terapeutica di prima scelta, soprattutto in presenza di microadenoma ipofisario (con diametro cioè, inferiore ad 1 cm). In questo caso, infatti, i risultati della neurochirurgia sono buoni, con percentuale di guarigione di circa 80%. In presenza di macroadenoma (evenienza assai più frequente) l’efficacia della terapia neurochirurgica è assai più modesta, con percentuale di guarigione mai superiore al 40% dei casi.
La radioterapia è stata per lungo tempo considerata l’opzione terapeutica di seconda scelta e generalmente proposta nelle situazioni in cui le condizioni del paziente non consentano l’intervento o se il tumore è profondamente infiltrato a livello della regione ipotalamica. La radioterapia può essere effettuata con metodica convenzionale, frazionata a campi contrapposti o stereotassica, se dimensioni e sede della neoplasia lo consentono. Il limite principale di quest’opzione terapeutica è rappresentato dall’efficacia assai tardiva e dalle complicanze. Infatti una normalizzazione della secrezione di GH e IGF-I viene spesso riscontrata solo dopo dieci anni dall’applicazione ed è quasi sempre associata a ipopituitarismo. Per questo motivo, la radioterapia classica viene attualmente sconsigliata da molti autori, soprattutto a fronte di efficaci opzioni di terapia medica.
Per quanto riguarda la terapia medica, i farmaci storicamente utilizzati nel trattamento dell’acromegalia sono stati i dopamino-agonisti, quali bromocriptina e cabergolina, e, soprattutto, la somatostatina ed i suoi analoghi, in particolare nelle loro formulazioni ritardo.
Attualmente, il trattamento farmacologico di scelta si basa su analoghi della somatostatina quali octreotide e lanreotide, che sono disponibili in formulazioni ad attività ritardata, efficaci fino ad un mese dopo singola somministrazione. Questi analoghi della somatostatina possiedono uno spiccato effetto inibitorio sulla secrezione di GH; a quest’azione fa seguito una riduzione dei livelli di IGF-I circolanti. L’effetto inibitorio degli analoghi della somatostatina si esercita sulla secrezione ma non sulla sintesi di GH; tuttavia l’efficacia non si riduce durante trattamento cronico e si associa talvolta ad una riduzione delle dimensioni della neoplasia ipofisaria. La terapia con somatostatina è particolarmente indicata dopo intervento neurochirurgico e/o radioterapia che non abbiano determinato guarigione, oppure, in prima istanza, in pazienti con controindicazioni alla terapia chirurgica e/o radiante. Peraltro tale terapia viene spesso prescritta anche in attesa dell’intervento, sulla base di segnalazioni (mai confermate) che ipotizzano un miglioramento del risultato chirurgico, oltre che delle condizioni generali, a seguito dell’inibizione dell’ipersecrezione di GH e IGF-I.
La reale normalizzazione della secrezione di GH e IGF-I in pazienti acromegalici durante terapia con analoghi della somatostatina non supera il 60% dei casi. Per i pazienti in cui la terapia farmacologica con analoghi della somatostatina si rivela inefficace nel normalizzare le concentrazioni di IGF-I o risulta non tollerata, sono attualmente disponibili nuovi farmaci, gli antagonisti recettoriali del GH, che bloccano l'attività dell'ormone della crescita, inclusa la produzione di IGF-I, nei tessuti periferici.
Tali farmaci risultano ben tollerati e sembrano normalizzare le concentrazioni di IGF-I in circa il 90% dei pazienti trattati.